|
Questions:
- How to increase the oligos Picky designed for my genome?
- In Picky 1.1 and newer, the closest nontarget melting temperature does not seem to match the one shown to me when I double-click on the oligo to bring up the detail view windows. Is that a bug?
- For genome "tiling" purpose, how to use Picky to find all oligos that are either unique in the whole genome or are shared only by a few locations on the genome, no matter whether they target a gene encoding region or not?
- I saved my input file as text file but when I try to load that into Picky it keeps saying "file contains no sequence data". Why?
- The temperatures reported in Picky windows are way high; most microarray experiments were conducted at a much lower temperature. Is this an error?
- In the Picky oligo output file, there is a new Type code field associate with each identified target and nontraget. What does the type code mean?
- Picky 2.20 adopted a new output file format. Could you briefly explain what the output contains?
- I am excited about the new oligo examination feature in Picky 2.1, and I want to check my existing array oligos to see how good they are. What is the file format of the oligo input (in contrast to regular sequence input)?
- I ran Picky on my genome sets but it crashed or was stuck in step 1 "suffix array construction" for many hours. Is that a bug?
- I ran Picky on third party oligos to check their quality, but I saw some oligos were shown with mixed colors of black, blue and green. What does that mean?
- I ran Picky to check the vendor designed oligos against my gene sets, but Picky found very few matches. How's that possible because I know the array can't be that bad?
- Picky on my computer is displaying an ugly font for DNA bases. How do I adjust the font used by Picky?
- What is the nontarget gene set; when and how to use the nontarget gene set?
- What are the main differences between the 32-bit Picky and 64-bit Picky?
Q1: How to increase the oligos Picky designed for my genome?
A: There are several options you can try to increase the number of oligos designed by Picky:
a. Switch to another genome. Some genomes are highly repetitive due to transposons and other artifacts. If you can choose to work on another genome you may have a higher chance to get more oligos, although this may not be a good alternative for most people. :)
b. Try to increase the maximum match length. The maximum match length parameter in Picky determines sequence regions that will not be considered further for oligo design because they are too similar to each other. However, for large genomes, by random chance many regions will be covered by a few short overlaps that jointly kill the regions. If this parameter is increased slightly it will admit more regions for oligo considerations and increase the total number of oligos designed. However, it will also increase Picky processing time.
c. Try to lower the minimum temperature separation parameter. If an oligo candidate has a close nontarget, its melting temperature with the nontarget may not be too different from its melting temperature with its own target. By default, Picky will toss away oligo candidates whose target/nontarget temperature difference are less than 15 degree Celsius. You can lower this limit to increase the number of oligos selected. However, if you set this parameter too low you run the risk of cross-hybridizations.
d. Try to use the oligo sharing feature in Picky 2.0 or later. The fact is that many large genomes just can't have unique oligos designed for every gene family members due to their high similarity. It is thermodynamically impossible to separate them with hybridization temperature differences. However, a single oligo can sometimes be shared by an entire family to detect the presence of any family member, even if they cannot be individually identified.
Q2: In Picky 1.1 and newer, the closest nontarget melting temperature does not seem to match the one shown to me when I double-click on the oligo to bring up the detail view windows. Is that a bug?
A: When Picky is conducting the nontarget melting temperature calculation, all detected nontargets are aligned with the target sequence and the best oligo probes are designed. Note that the same nontarget sequence can usually have multiple local matches and alignments with the target, and only its highest match temperature was recorded for that nontarget in the oligo panel. However, in the detail probe region overlap view, only nontarget matches overlapping the same probe target region will be displayed and whose melting temperature dynamically estimated. This can be lower than the highest recorded nontarget melting temperature whose alignment was initiated outside of the probe target region. An example of this can best illustrate the situation:
If you run Picky 2.0 on the dual.seq, you will find the closest nontarget temperature in the Oligo Probes panel being higher than the estimated temperature for Contig 2167 in the detail probe target region overlaps view.
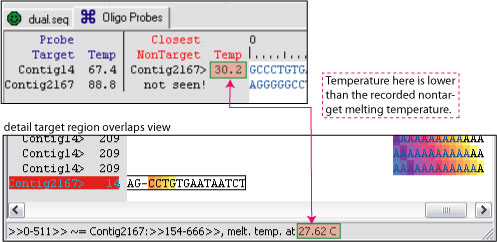
However, if you double click Contig 14 in the dual.seq panel, it will bring up the general longest overlaps view. Here you will see Contig 2167 having multiple local matches and alignments with Contig 14. The one with the gap to the right of CC produced the highest estimated 30.2 degree shown in the oligo panel, but the one with the gap to the left of CC was the one used to estimate and arrived at the 27.62 degree estimate in the overlap view window.
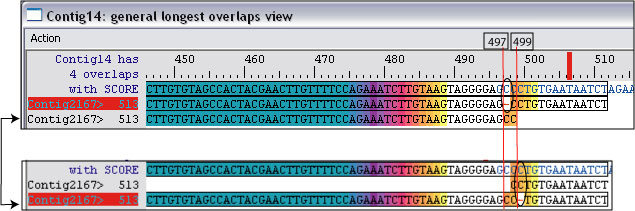
Generally, the dynamically estimated melting temperature should be the same or lower than the highest temperature recorded in the oligo panel for each nontarget. Note that Picky 2.1 now comes with an extra step to recompute oligo probe temperatures with its nontargets from the perspective of the probes themselves. Therefore, this is less likely to happen in Picky 2.1 or later.
Q3: For genome "tiling" purpose, how to use Picky to find all oligos that are either unique in the whole genome or are shared only by a few locations on the genome, no matter whether they target a gene encoding region or not?
A: There are two Perl scripts included here for designing genome tiling oligos using Picky. The break_up.pl file will break up large genomic sequences into smaller chunks that overlap a user specified length. Once you use this script to split up your input sequences, you can use Picky to design oligos.
After Picky oligo design, use the merge_back.pl script to update oligo coordinates against the whole genomic sequence and to consolidate oligos that might have been designed against an overlapping area, thus was considered by Picky as shared oligos with duplicative output entries.
The merge_back.pl script ignores nontargets and oligo types (U or S) since these are of auxiliary use only. To make them also correct we would need to record the original genomic sequence length and to check if a target is still unique after merging, etc. Those enhancements are left to the users.
If you use Windows computers you can download Perl executable from www.activeperl.com.If you use Unix or Mac you should have Perl already installed on your computers. On all three platforms, however, you need to run the two scripts in a command terminal window with some arguments. Look into the content of the two Perl program files or just run them without any argument to see a brief description about their usage.
Q4: I saved my input file as text file but when I try to load that into Picky it keeps saying "file contains no sequence data". Why?
A: Since you did not specify the platform where you are running Picky, it's not easy to pinpoint the exact problem. However, you should know the following two basic requirements:
1. Picky reads strictly FASTA format DNA sequences in pure text files (ASCII files) ONLY! Your file input should look like this when viewed in a "text editor", and note that Microsoft Word is NOT a text editor:
:
>DNA1
ACTG.... several lines
>DNA2
ACGT.... several lines ...
2. It doesn't matter what the file extension says (.seq, .txt, etc.); you still need to make sure its *content* is valid, not just its file name extension. To check the actual content of your input file for correctness, do one of the following depending on your platform:
On a PC platform, use the "Notepad" program to read your file. If you can't see its content correctly, Picky can't see your data either.
On a Mac platform, use the "TextEdit" program to read your file. If you can't see its content correctly, Picky can't see your data either.
On a Unix/Linux platform, use the "less" program for the same check.
Note that you should avoid loading your data back into Microsoft Word or any other *word processor* since they sometimes add invisible characters to a text file to keep their own book-keeping record (date of modification, ownership, etc) and those hidden invisible data can screw up Picky reading.
Finally, check your input file size and make sure it does not show zero bytes; otherwise indeed your file contains no sequence data!
Q5: The temperatures reported in Picky windows are way high; most microarray experiments were conducted at a much lower temperature. Is this an error?
A: Not exactly. Picky is conducting its calculation using the best known nearest neighbor thermodynamics parameters obtained by biochemists in aqueous buffers containing only water and salt, but most microarray array experiments were conducted in semi-aqueous phase with one partner of the hybridization, namely the probes, fixed to a solid surface. Currently, there are no known thermodynamic parameters to directly estimating the DNA annealing characteristics on microarray surfaces. Even if such parameters exist, they likely will be platform and protocol dependent and may not be generally applicable to other microarrays and protocols.
What Picky does is to estimate the melting temperatures of oligos with their targets or nontargets as if they are free floating in aqueous buffers, with total degree of freedom to move around and with all bases of the oligo participating in hybridizations. Picky then identifies the best oligos and their closest nontargets under such idealized conditions. This is the most general way to oligo design since it is platform independent and uses the best known thermodynamic parameters. Granted, this does not reflect the actual conditions on microarray chips and under specific hybridization protocols. Therefore, any lab using Picky designed microarrays needs to conduct their own calibration experiments using their spike controls or negative controls on the arrays to arrive at a suitable microarray experiment temperature. To summarize, the Picky reported temperatures should be interpreted only as a gauge of the likelihood of cross-hybridizations among oligos, targets and nontargets. The basic assumption is that even the actual temperatures under microarray experiments will be different than what Picky reported, the shift toward the real ones will be linear or close to linear, meaning that good oligos will still be good oligos, just that you need to observe your own estimated optimal experiment conditions to make the best of them.
In order to assist scientists calibrating their microarrays designed by Picky, we are currently working on a quantitative optimal microarray hybridization temperature calibration protocol. This new protocol will be able to empirically determine the actual shift in melting temperatures from Picky determined theoretical values toward the protocol and array dependent actual temperatures. Once this protocol is published, a new Picky 2.2 will be released which can then take in the calibration results and automatically adjust its reported temperatures to match those in actual experiment conditions. Stay tuned for this new calibration protocol and Picky 2.2!
Q6: In the Picky oligo output file, there is a new Type code field associate with each identified target and nontraget. What does the type code mean?
A: Because of the new shared oligo design capability introduced in Picky 2.0, the .picky output file format has been modified somewhat. Most fields should be self-explanatory from its first title line. Only a new 'Type' field has been created to remark on the nontarget or target gene names for each oligo:
1. Type code U means a gene has unique oligos. Note that it does not mean a gene with this Type code cannot be listed on the shared oligo lines. Of course for all unique oligos their targets should carry this Type code only, but shared oligos sometimes also target these genes due to some unavoidable conflicts with Type code S genes;
2. Type code S means a gene has only shared oligos, i.e., it cannot be uniquely identified with the oligos. A shared oligo should target at least one gene having this Type code for it to be chosen, but as mentioned above it might also target some other genes that have their own unique oligos already listed earlier in the Picky output;
3. Type code R means the user has reverse-complemented an input sequence before starting the oligo computation. To maintain consistency, Picky always uses coordinates related to the original input sequence orientation. This is usually not relevant to users since most people do not need to reverse-complement their input sequences, or have already done so before loading the data into Picky;
4. Type code > means a probe matches a nontarget in its forward strand; and
5. Type code < means a probe matches a nontarget in its reverse strand.
Type code can be combined, e.g., 'R >' means a reverse-complemented nontarget forward strand, and 'R U' means a reverse-complemented target that has unique oligos. All data fields are separated by the tab (\t) characters as before, and within the Type code fields the code are separated by one space character for easy parsing by Perl.
Q7: Picky 2.20 adopted a new output file format. Could you briefly explain what the output contains?
A: The new Picky 2.20 output format is block oriented, not line oriented as in the previous output format. The new output format is more versatile and allows multiple targets and nontargets to be reported for each probe. It is also much easier to read or to be parsed by Perl code.
The content of each data block is related to one designed or examined probe; data on each line are separated by the TAB character:
First line: probe sequence, probe length, No. of targets reported, No. of nontargets reported, lowest target melting temperature, and highest nontarget melting temperature
Subsequent lines: Type code (U: unique, S: shared, >: nontarget on forward strand, <: nontarget on reverse strand; all of these can be augmented with the R code to mean that the input sequence has been reverse-complemented), melting temperature with the probe, probe annealing site left coordinate, probe annealing site right coordinate, target or nontarget annealing site left coordinate, target or nontarget annealing site right coordinate, and the target or nontarget gene name.
Last line: blank to separate a block from the next one.
Q8: I am excited about the new oligo examination feature in Picky 2.1, and I want to check my existing array oligos to see how good they are. What is the file format of the oligo input (in contrast to regular sequence input)?
A: Free format, simply put. While regular sequence input to Picky is required to be in the FASTA format, oligo input to Picky for examination purpose can be in almost any text file format. Picky will search within the oligo input files for anything that looks like an oligo, i.e., is continuous pure A, C, G, T or U code and within the maximum and minimum oligo lengths limit. Therefore, you should set your maximum and minimum oligo lengths parameter to match your oligos. This is why Picky can directly read its own .picky files back despite that in these files there are many other non-oligo data. If there are other non-oligo DNA data that happen to be within the length limit set for the oligos, Picky will read them as oligos. Therefore, if this might happen to your input you should clean up your oligo files before loading them into Picky for examination.
Q9: I ran Picky on my genome sets but it crashed or was stuck in step 1 "suffix array construction" for many hours. Is that a bug?
A: If Picky crashes or was stuck in step 1 or 2, then most likely you are simply running out of free memory on your computer to construct the suffix, invert and LCP arrays. 32-bit Picky needs about 20 times your input gene set size memory to conduct its computation. 64-bit Picky needs even more at 40 times your input gene set size memory to conduct its computation. For example, if you have a 2G memory computer, you may have just about 1.8G free memory for applications since the operating system will reserve some memory for itself (could be even lower if you also run other applications alongside Picky), and therefore you cannot load gene sets with a combined size more than 1.8G/20 = 90M into 32-bit Picky and expect Picky will finish its computation efficiently.
For 32-bit computers, the maximum memory space that can be addressed by a single program like Picky is 4G, theoretically. However, operating systems always put some additional limit on it, thus in reality most 32-bit computers cannot have more than 3G free memory for Picky, even on a 4G memory computer! That puts an upper limit of about 150M on the maximal gene set(s) that can be processed by 32-bit Picky. To process even bigger gene sets beyond 150M, you will need 64-bit Picky and run it on 64-bit computers with lots of memory.
Q10: I ran Picky on third party oligos to check their quality, but I saw some oligos were shown with mixed colors of black, blue and green. What does that mean?
A: It means that some third party oligos overlap each other on the same gene target region. Since each sequence base in Picky can assume just one color, when oligos overlap each other on their target, Picky can only paint the target bases with one chosen color. The order of color choices is black, blue and green with green (i.e. shared) oligos having the highest precedence if the same location was also targeted by black (i.e., nonspecific) and blue (i.e., unique) oligos. This also means that double-clicking in the sequence display may not get you the correct oligo targeting region overlap view because the colors do not reflect the oligo size and Picky has no way of knowing which particular oligo's view you want to see. You will have to do that double-clicking in the probes panel directly.
Since Picky's own oligos never overlap each other, therefore there is no better way to display the overlapping oligo information when checking third party oligos. To find out which third party oligos overlap each other, you will have to save the whole oligo check results and use a Perl script to identify them.
Q11: I ran Picky to check the vendor designed oligos against my gene sets, but Picky found very few matches. How's that possible because I know the array can't be that bad?
A: Please reverse-complement your gene sets and try again. If you can see many matches now, then likely that's just an orientation problem. Picky assumes all designed oligos are in the same read direction as its input gene sets, i.e., the oligos are actually targeting the opposite strands of the input gene sequences. If the vendor oligos are designed to target the same strands, then Picky will not find them. In fact, even if Picky find some matches it will consider them bad oligos because they may target areas that can form secondary structures. There are many different microarray hybridization protocols, and some also involve the amplification of RNA, resulting in different orientations of the dye-labeled final transcripts to be hybridized against the arrays. If you purify your mRNA and use direct reverse-transcription with dye incorporation in one step to obtain your final labeled transcripts, then they will be in the anti-sense orientation and the array oligos should be on the sense strands (i.e., same as gene reading direction) in order to hybridize with them. If you use a T7 aRNA amplification step like those provided by Ambion, your amplified RNA will be in the anti-sense strand and the final labeled transcripts will be in the sense strand. Contrarily, if you use a sense-strand RNA amplification kit like those provided by Genisphere, your amplified RNA will be in the sense-strand and the final labeled transcripts will be in the anti-sense strand, same as in the non-amplified protocol. Anyway, just be sure you match the final labeled transcripts with the oligos on the array, so you will know if they can hybridize with each other to form double-stranded complexes.
Q12: Picky on my computer is displaying an ugly font for DNA bases. How do I adjust the font used by Picky?
A: Instead of trying to adjust the fonts used by Picky, please try to locate the fonts required by Picky. Believe us, we have tried many different fonts on different computing platforms and arrived at the best fonts to display DNA bases for each platform: On Linux Picky will use "Lucida Typewriter", on Windows Picky will use "Courier New", and on Mac Picky will use "Andale Mono" fonts . If the font specific to your platform is not available on your computer, then Picky will be given a system default font which may not look good. These fonts we mentioned should come standard with these three platforms, but you may have accidentally deleted them or chosen not to install them. It is beyond the scope of this FAQ to tell you how to locate and re-install these fonts for your computing platforms. Please find your local computer guru to help you ensure that Picky can have access to these fonts, and then your DNA and oligo sequences should look good on screen.
Q13: What is the nontarget gene set; when and how to use the nontarget gene set?
A: The nontarget gene set is used when you want to prevent Picky from designing oligos
that might be confused by other sequences "during the same
microarray experiment". Note that only sequences that may potentially influence
your experiment outcome need to be screened by loading them into the
nontarget set. Usually, if you are
conducting microarray experiments on mRNAs, you do not need to
screen against the whole genome since your only concern is if one mRNA
might be mistaken as another mRNA by Picky designed oligos. When designing a whole genome array, ALL your coding mRNAs are in
the "target" gene set already , so you got nothing in the "nontarget" set . This may not be
true, for example, if you do NOT have a mRNA purification step in your array
protocol before labeling them. In that case, you would probably put
tRNA, rRNA, siRNA, miRNA and any other non-coding RNA transcripts that
"might be there" into the "nontarget" set. Although, in general, to
improve your experiment results you would want an mRNA purification
step for eukaryotes.
If you are just targeting a few genes for oligo design, then you do need to put the
rest of the genes into the nontarget set. You need to separate
your genes into two sets, so the same sequence will *not* show up both
in the target and nontarget sets. If it does, it will NOT have any
oligo designed. You may be asking why Picky does not automatically
mask the sequences found in the target set from the nontarget set, if
they were found there at the same time. There are many reasons why
Picky does not make this "same sequence" call for its users:
1. Those might be paralogs, not the same genes, and Picky cannot tell
one from the other just because they are the same;
2. It wastes computer memory to store the same sequence multiple
times, and it also slows down Picky;
3. It is too much trouble to implement this nonessential "feature" into the
already very complex Picky algorithms; and
4. It's SO easy to separate the genes ahead of time before you run
Picky, instead of mixing them in and hoping that Picky will "do the
right thing".
Your remaining question might be then how do I separate my source gene sequences into two sets,
the target and nontarget. Well, if you know Perl this is very easy; here is a sample Perl program select.pl that will select sequences
given in its command-line arguments into a new set. If, however, you
do not know any Perl programming at all, you will need to find some
local bioinformaticists to help you separate your gene sets.
Q14: What are the main differences between the 32-bit Picky and 64-bit Picky?
A: Functionally, they are exactly the same since they are compiled from the same source code. The main differences lie in their data processing capability limits. For all practical applications, the 64-bit Picky is considered unlimited given enough computer memory. However, 64-bit Picky uses larger addressing space, thus requires more memory overhead to process the same amount of input data. If you have less than 8G main memory, then 32-bit Picky running on a 64-bit operation system (if configured to support 32-bit code) will be the best choice. The following table listed the main differences between the two types of Picky.
Data limits |
32-bit Picky |
64-bit Picky |
Remarks |
| Maximum gene sets |
2 |
256 |
Target and nontarget gene sets are counted together. |
| Maximum number of sequences per gene set |
65,532 |
16,777,212 |
Gene sets larger than this limit can be split into two but the maximum gene sets apply. |
| Longest allowable input gene sequence length |
16,384 |
2,147,483,647 |
When longer than the longest length, Picky can break up the sequence into multiple parts. However, the total gene count will be increased this way. |
| Shortest allowable input gene sequence length |
50 |
50 |
Input sequences shorter than this can be skipped by Picky. |
| Memory overhead for input data |
20X |
40X |
The amount of extra memory needed during computation. |
| Maximum number of computing threads |
1024 |
1024 |
The available number of threads Picky can actually use is usually higher on 64-bit machines. |
In addition to the differences in capabilities, the licensing agreements for 32-bit and 64-bit Picky are also different. We try to make Picky as widely available as possible, so only commercial users licensing 64-bit Picky for commercial use will actually need to pay a fee.
User category |
32-bit Picky |
64-bit Picky |
| Academic or nonprofit users using Picky for nonprofit purposes |
|
|
| Industrial or commercial users using Picky for commercial purposes |
|
|
|

